

Getting Started
BUSCO runs on the command line, using a Terminal or Console application. You should have some familiarity with the basics of working with the command line.
BUSCO is a software package that requires a number of third-party tools. To simplify installation, BUSCO is available as a Conda package and a Docker image with all dependencies pre-installed. It can also be installed manually though in this case care must be taken to ensure all dependencies are installed and configured correctly.
Installation with Conda
Important Notice: Auto-lineage Compatibility
Some users may experience unexpected lineage assignments when running BUSCO with SEPP v4.5.6 (this may concern users who installed BUSCO v6.0.0 from Conda before Dec. 2).
This is due to a compatibility issue between the auto-lineage files and this newer version of SEPP.
The last BUSCO v6.0.0 on Conda pins SEPP to v4.5.5 by default. Make sure your installation is using SEPP 4.5.5 for auto-lineage compatibility, and reinstall from Conda if needed.
(For the Docker image, no action is required).
Conda is an open-source package management and environment management system that simplifies the installation and management of software packages and their dependencies across diverse computing environments.
Ensure sure you have conda version 4.8.4 or higher. Enter
conda -Vto check. If necessary, update conda by enteringconda update -n base conda.To install BUSCO in the current environment, enter
conda install -c conda-forge -c bioconda busco=6.1.0Alternatively you can create a new environment with BUSCO installed
conda create -n <your_env_name> -c conda-forge -c bioconda busco=6.1.0 conda activate <your_env_name>
If conda takes a long time to solve the environment, you can try using mamba instead of conda. Mamba is a reimplementation of the conda package manager in C++.
conda install -c conda-forge mamba
mamba install -c conda-forge -c bioconda busco=6.1.0
Installation with Docker
Docker is a tool for packaging and running applications and their dependencies in isolated containers, streamlining the deployment and management of software across diverse environments. Some familiarity with Docker is required to use the BUSCO Docker image. See the Docker userguide for details.
In short
docker pull ezlabgva/busco:v6.1.0_cv2
docker run -u $(id -u) -v $(pwd):/busco_wd ezlabgva/busco:v6.1.0_cv2
Avoid running containers in root, specify your user uid
docker run -u $(id -u) ezlabgva/busco:v6.1.0_cv2
Use mounts (-v) to exchange files between the host filesystem and the container filesystem. In the following example your current working directory ($(pwd)) is mounted as /busco_wd in the container. This allows you to access files in the current directory from within the container and to write files to the current directory from within the container.
docker run -u $(id -u) -v $(pwd):/busco_wd ezlabgva/busco:v6.1.0_cv2
The default working directory in the container is /busco_wd. With your non-root uid, attempts to write in the container virtual filesystem will fail if it is not a mount point. You can redefine the working directory using -w to match another mounted folder as current working directory.
For example, if your BUSCO input filepath on your file system is /home/name/genome.fna
docker run -u $(id -u) -v /home/name/:/busco_wd ezlabgva/busco:v6.1.0_cv2 busco -i genome.fna
is equivalent to
docker run -u $(id -u) -v /home/name:/host_mount -w /host_mount ezlabgva/busco:v6.1.0_cv2 busco -i genome.fna
Be careful not to specify a host folder that does not exist when using -v /hostfolder/:/dockermount. Docker will create it with the root account, which is useless and annoying. It is safer to use the current directory -v $(pwd):/dockermount
Manual installation
To do a manual installation you will need to install the BUSCO code and all of its third-party dependencies separately. This is not recommended unless you have a specific reason to do so.
Installing BUSCO code
The following steps should be performed in a dedicated environment. See here for more on virtual environments.
- Clone the repository.
git clone https://gitlab.com/ezlab/busco.git cd busco/ - Install using pip from within the cloned directory.
python -m pip install .NB: We recommend using a virtual environment to avoid conflicts with other Python packages.
Third-party components
A full installation of BUSCO requires
- Python 3.3+ (2.7 is not supported from v4 onwards)
- BioPython
- Pandas
- BBMap for genome assembly statistics such as N50
- Miniprot default for eukaryotic genome and eukaryotic transcriptome modes
- tBLASTn 2.2+ for eukaryotic genome and prokaryotic transcriptome modes
- Augustus 3.2+ option for eukaryotic genome mode
- Metaeuk option for eukaryotic genome and eukaryotic transcriptome modes
- Prodigal for prokaryotic genome mode
- HMMER3.1+ for all modes
- SEPP for the auto-select lineage pipeline. IMPORTANT: Use SEPP version 4.5.5. SEPP v4.5.6 is incompatible with BUSCO autolineage files and results in wrong lineage assignments.
Please make sure that each software package listed above works INDEPENDENTLY of BUSCO before attempting to run any BUSCO assessments.
Relevant parameters given to BUSCO are propagated to third-party tools (e.g. number of cpus). Their default configuration should not be changed for BUSCO runs.
For Augustus, the option --augustus_parameters allows advanced users to freely pass parameters. Use it only to fix biologically relevant parameters such as the translation table and mention these when reporting the result. Do not use it for selecting the Augustus species, as there is a dedicated BUSCO parameter (--augustus_species), and do not use it to specify the number of cpus (-c, --cpu).
Augustus
Augustus uses several executables and PERL scripts. Please refer to Augustus documentation for PERL requirements.
Augustus requires environment variables to be declared as follows:
export PATH="/path/to/AUGUSTUS/augustus-3.5.0/bin:$PATH"
export PATH="/path/to/AUGUSTUS/augustus-3.5.0/scripts:$PATH"
export AUGUSTUS_CONFIG_PATH="/path/to/AUGUSTUS/augustus-3.5.0/config/"
NB: you can use the printenv command to view all your environment settings.
Known bugs and unsupported versions
tBLASTn 2.4+
During development, we recognised an issue with tBLASTn versions 2.4-2.10.0 when using more than one CPU. NCBI issued a fix for the problem in version 2.10.1+. Make sure you have at least version 2.10.1+ installed.
Supported OS
BUSCO is being developed and tested on multiple distributions of Linux (e.g. Arch Linux, CentOS, Ubuntu). We do not support macOS but BUSCO should work on it, although Augustus seems to cause troubles on some BSD-derived systems, including macOS. Consider the Docker container if you work on incompatible environments.
Running BUSCO
Command Line Options
Mandatory parameters
busco -i [SEQUENCE_FILE] -m [MODE] [OTHER OPTIONS]
-i or --in defines the input file to analyse which is either a nucleotide fasta file or a protein fasta file, depending on the BUSCO mode. As of v5.1.0 the input argument can now also be a directory
containing fasta files to run in batch mode.
-m or --mode sets the assessment MODE: genome, proteins, transcriptome
Recommended parameters
-l or --lineage_dataset Specify the name of the BUSCO lineage dataset to be used, e.g. kitasatospora_odb12.2. A full list of available datasets can be viewed by entering busco --list-datasets.
You should always select the dataset that is most closely related to the assembly or gene set you are assessing. If you are unsure, you can use the --auto-lineage option to automatically select the most appropriate dataset.
BUSCO will automatically download the requested dataset if it is not already present in the download folder. You can optionally provide a path to a local dataset instead of a name, e.g. -l /path/to/my/dataset.
-c or --cpu Specify the number of threads/cores to use. Unless this is specified BUSCO will only use one CPU, which could cause a long run time.
-o or --out Give your analysis run a recognisable short name. Output folders and files will be labelled with this name. If not specified the output will take the form "BUSCO_<input_filename>"
Optional parameters
--augustus Use augustus gene predictor for eukaryote runs.
--augustus_parameters "--PARAM1=VALUE1,--PARAM2=VALUE2" Pass additional parameters to Augustus. See Augustus documentation for parameter options. All parameters should be contained within a single string with no white space, with each parameter separated by a comma.
--augustus_species AUGUSTUS_SPECIES Specify a species for Augustus training.
--auto-lineage Run auto-lineage pipeline to find optimum lineage path.
--auto-lineage-euk Run auto-placement just on eukaryota tree to find optimal lineage path.
--auto-lineage-prok Run auto-lineage just on prokaryota trees to find optimum lineage path.
--config /path/to/config.ini Provide a config file as an alternative to command line parameters.
--contig_break n Number of contiguous Ns to signify a break between contigs in BBTools analysis. Default is n=10.
--download [dataset ...] Download dataset. Possible values are a specific dataset name, "all", "prokaryota", "eukaryota", or "virus". If used together with other command line arguments, make sure to place this last.
--download_base_url DOWNLOAD_BASE_URL Set the url to the remote BUSCO dataset location.
--download_path DOWNLOAD_PATH Specify local filepath for storing BUSCO dataset downloads. The default is a busco_downloads subdirectory in the current working directory.
-e N, --evalue N E-value cutoff for BLAST searches. Allowed formats, 0.001 or 1e-03 (Default: 1e-03).
-f, --force Force overwriting of results folder if it already exists. To be used with caution, as any previous results will be lost.
-h, --help Show this help message and exit.
--limit N How many candidate regions (contig or transcript) from the BLAST output to consider per BUSCO (default: 3). This option is only effective in pipelines using BLAST, i.e. the --augustus genome pipeline and the prokaryota transcriptome pipeline.
--list-datasets Print the list of available BUSCO datasets.
--long Optimize Augustus self-training mode. This adds considerably to the run time, but can improve results for some non-model organisms.
--metaeuk Use Metaeuk gene predictor.
--metaeuk_parameters "--PARAM1=VALUE1,--PARAM2=VALUE2" Pass additional arguments to Metaeuk for the first run. See Metaeuk documentation for parameter options. All parameters should be contained within a single string with no white space, with each parameter separated by a comma, e.g. --metaeuk_parameters="--max-overlap=15,--min-exon-aa=15"
--metaeuk_rerun_parameters "--PARAM1=VALUE1,--PARAM2=VALUE2" Pass additional parameters to Metaeuk for the second run. See Metaeuk documentation for parameter options. All parameters should be contained within a single string with no white space, with each parameter separated by a comma.
--miniprot Use Miniprot gene predictor (default for eukaryota in genome mode).
--offline In offline mode BUSCO will not attempt to download files. Ensure all required dataset files are already downloaded and available.
--opt-out-run-stats Opt out of data collection (from v5.6.0). Collected data is used to improve BUSCO. All collected data is anonymised and includes the pipelines used, the datasets selected, options used and runtime statistics.
--out_path OUTPUT_PATH Optional location for results folder, excluding results folder name. The default is the current working directory.
--plot DIRECTORY Create a BUSCO plot of results in the specified directory.
--plot_percentages For use with --plot. Plot values as percentages instead of numbers of marker genes.
-q, --quiet Disable the info logs, displays only errors.
-r, --restart Continue a run that had already partially completed. Restarting skips calls to tools that have completed but performs all pre- and post-processing steps.
--scaffold_composition Writes ACGTN content per scaffold to a file scaffold_composition.txt. Used by BBTools.
--skip_bbtools Skip BBTools for assembly statistics.
--tar Compress some subdirectories with many files to save space.
-v, --version Show this version and exit.
Editing BUSCO Run Configuration
BUSCO will try to use the dependencies available in your environment. For the Conda and Docker distributions these are pre-installed.
For the manual installation, it is up to the user to ensure the dependencies are available.
An alternative to making the tool commands available in your local environment PATH is to point to the locations of the tools in a BUSCO configuration file, config.ini.
A config file template is available here.
This can also be used to override the PATH if you have a reason to use a specific version of a third-party tool.
All command line options can also be specified in the config file instead of on the command line.
BUSCO will then first attempt to find the tools specified in the config file before falling back on the environment PATH variable.
Providing input parameters through the command line will override those defined in config.ini.
To activate this config file, either pass it as a command line option
busco --config /path/to/myconfig.ini
or set the environment variable BUSCO_CONFIG_FILE with the path to the file, as follows:
export BUSCO_CONFIG_FILE="/path/to/myconfig.ini"
Tips for running BUSCO
- Generally the lineage to select for your assessments should be the most specific lineage available, e.g. for assessing fish data one would select the actinopterygii lineage rather than the metazoa lineage.
A full list of available BUSCO datasets can be obtained by entering
busco --list-datasets
- If you are assessing a large number of species/strains/versions etc. then to minimise runtime (at the expense of resolution) one might select a less specific lineage set with fewer BUSCOs, e.g. for assessing 20 bird genomes each with a couple of different assembly versions one might select the vertebrata or the metazoa lineages rather than the aves lineage, at least for the initial rounds of assessments.
- Assessments generally produce several folders with lots of files (see BUSCO pipelines). These are for your benefit, so that you can examine individual cases in more detail and/or use the data for downstream analyses.
- Please do take some time to check the log files, these are there for your benefit in order to highlight potential problems that may have occurred during your BUSCO run.
- Compare the results from assessing your data with like-for-like assessments of corresponding publicly available data for other closely-related species. In this way, the BUSCO results can be used to claim that your dataset is as good as, or better than, existing publicly available datasets for similar species.
- If manual curation of annotated gene sets was performed, report BUSCO results before and after curation to quantify improvements.
Lineage datasets
BUSCO uses a set of evolutionarily-informed expectations of gene content of near-universal single-copy orthologs to assess the completeness of genome assemblies, gene sets, and transcriptomes. These expectations are derived from the analysis of a large number of evolutionarily diverse species, and are provided in the form of lineage-specific datasets. Marker genes are selected as those that are present in at least 90% of the species in a given lineage, and present in a single copy in 90% of those species. The marker genes are used to assess the completeness of the genome assembly or gene set being analysed. BUSCO scores are reported as the percentage of these marker genes that are found in the assembly or gene set.
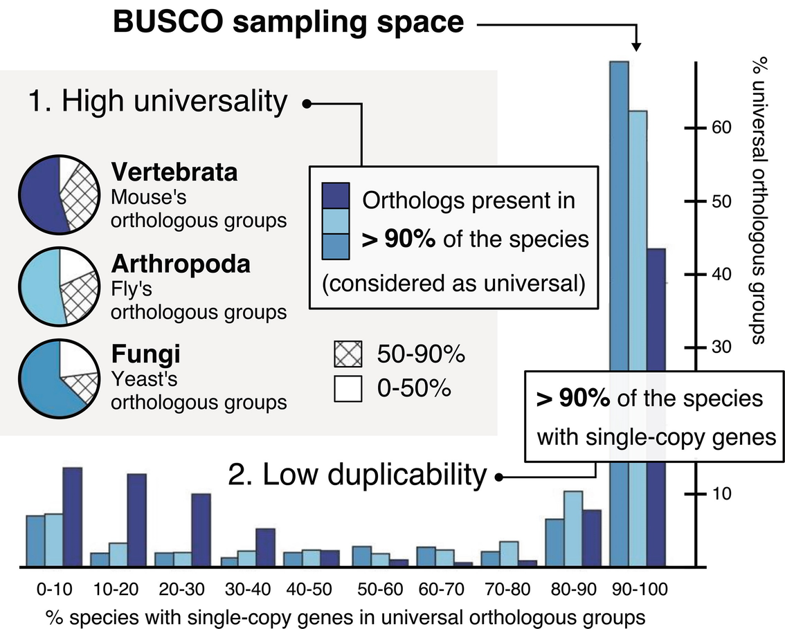 adapted from Waterhouse et al. (2010)
adapted from Waterhouse et al. (2010)
BUSCO provides a set of pre-computed datasets for a wide range of organisms. Any use of these datasets for analyses in a publication or product must include the citation of the corresponding paper: https://doi.org/10.1093/molbev/msab199.
Specify a lineage dataset with the -l or --lineage_dataset option, for example
busco ... -l enterobacterales_odb12.2 ...
BUSCO will automatically download the requested dataset if it is not already present in the download folder. You can optionally provide a path to a local dataset instead of a name, e.g. -l /path/to/my/dataset.
To print the full list of available datasets enter busco --list-datasets
For metagenomic analysis it is possible to let BUSCO selected automatically the datasets that is best suited to the input assembly, using the --auto-lineage, --auto-lineage-prok, or --auto-lineage-euk options.
The datasets contain the following directory structure:
eukaryota_odb12.2
├── ancestral
├── ancestral_variants
├── dataset.cfg
├── hmms/
├── info/
│ ├── ogs.id.info
│ └── species.info
├── links_to_odb12.2.txt
├── prfl/
├── refseq_db.faa.gz
├── scores_cutoff
└── lengths_cutoff
HMM Profiles
The hmms/ directory contains the HMM profiles for the marker genes in the dataset. These are used by BUSCO to identify
the presence of these genes in the input assembly or gene set. Each profile corresponds to an orthologous group (OG) of
genes on OrthoDB. From all the OGs at a given taxonomic level, BUSCO selects those in which
the constituent genes are present in at least 90% of the species in that lineage, and present in a single copy in 90%
of those species. For large levels with more than 100 species this threshold is proportionally tightened.
Of all the genes in a given OG, BUSCO samples evenly across all lower taxonomic levels (genus, family, order, etc.) and employs a number of statistical quality metrics to ensure a high-quality and balanced representation of the diversity of the lineage. The sampled genes are then aligned and a profile HMM is built from the alignment.
The prfl/ directory contains the block profile file for each BUSCO OG, used in the Augustus pipeline.
Match thresholds
The scores_cutoff and lengths_cutoff files contain thresholds used for evaluating BUSCO matches.
The scores_cutoff file contains the minimum HMMER bit score for a match to be considered a valid BUSCO hit.
The lengths_cutoff file contains thresholds and margins for the lengths of the matches to be considered Complete,
Fragmented, or Missing. The construction and use of this file has evolved in the BUSCO pipeline, and the exact
parameters depend on the dataset version.
For odb10 datasets, the thresholds are based on the mean lengths and standard deviations of the lengths of the
genes in the OG.
For odb12 datasets, the lengths_cutoff file was removed, and the length threshold for each OG was 80% of
the profile length of the OG.
For odb12.2 datasets, the lengths_cutoff file was reintroduced. The file now contains, for each group, the median
length and the scaled median absolute deviation (sMAD). For a match to be considered Complete, the revised logic
requires the matched length to be greater than the smallest of the following:
90% of the median length, median - (2 x sMAD) (capped at 25% of the median length), or 80% of the HMM profile length.
The upper bound for a Complete match is 1.5 x median.
BUSCO is version-aware, and will use the logic appropriate to the dataset version. It is recommended to keep the BUSCO
version up-to-date to avail of the latest improvements, as, for example, the odb12.2 datasets can only be correctly
parsed by v6.1.0 or later.
Information files
The info/ directory contains files containing a list of all species sampled from OrthoDB to create the BUSCO OG,
along with a mapping of each OG to the corresponding gene sequences for these species.
Note: Clustering is done from scratch for each version of OrthoDB (with the exception of prokaryotic clusters remaining
unchanged from v12.1 to v12.2). Cluster IDs are therefore not retained across dataset versions, and there is no
guarantee that a v10 cluster is present in v12.2, given the development of the species selection and clustering
algorithms. Nevertheless, to attempt to map a v10 OG to a v12.2 OG, it may be possible to compare the BUSCO OG gene
constituents from the ogs.id.info files.
The links_to_odb12.2.txt file contains the annotations of the OGs in the dataset and url links to the corresponding
OGs in OrthoDB v12.2.
The odb12 and odb10 datasets contain the corresponding links to the correct versions of OrthoDB.
Pipeline-specific files
The ancestral file contains a consensus ancestral sequence for each BUSCO, used by BLAST in the
Augustus pipeline and the prokaryotic transcriptome pipeline.
These consensus sequences are obtained by using MMSeqs Linclust.
The ancestral_variants file contains up to 10 variant sequences for each BUSCO.
These are used to expand the BLAST search for BUSCOs that were not matched by the consensus sequence.
The refseq_db.faa.gz file contains the reference protein sequences for each BUSCO used by Miniprot and
Metaeuk. This contains all sequences used to create the HMM profiles, but they are de-duplicated, so there may be
sequences in the info/ogs.id.info file that are not present in the refseq_db.faa.gz file.
The missing_in_parasitic.txt file, where present, contains a list of genes present in lineages containing clades with
reduced parasitic genomes (e.g. fungi_odb10).
The dataset.cfg file contains configuration parameters used in the various pipelines. This file
also contains information on the number of species used and the number of OGs in the dataset.
Download and automated update
BUSCO can obtain the last version of the lineage datasets. If the name of a dataset is passed, e.g. -l bacteria_odb12.2, BUSCO will download it automatically. If a full path is given to BUSCO using -l /my/own/path/bacteria_odb12.2, this automated management will be disabled. Otherwise, files used during the automated lineage selection process are also automatically obtained by BUSCO.
By default, if a new version of a file is available, BUSCO will warn you. If you pass the argument --update-data, busco will replace the current file or folder with the up-to-date version and archive the previous one.
Individual datasets can also be downloaded using the busco --download command, e.g. busco --download bacteria_odb12.2. It is also possible to do a bulk download by using the following arguments in place of the dataset name: "all", "prokaryota", "eukaryota", or "virus". Please exercise restraint when using the "download" command - excessive use of this command will put a strain on the BUSCO server and may result in your IP address being blocked.
Offline
If you are running BUSCO in an environment that does not have access to the Internet, you can pass the --offline argument to prevent BUSCO from attempting any download. Make sure you have first downloaded all the datasets you will need using the busco --download command. The location of the download folder should then be passed using the command line flag: --download_path /path/to/datasets.
BUSCO Pipelines
The BUSCO output folder name is BUSCO_<input_filename> by default, but this can be changed by using the -o or --out option.
This directory will contain several files and directories, which will vary depending on which pipeline you are using.
Genome mode
The genome mode is used to assess the completeness of genome assemblies. It uses the --mode option set to genome. The input file should be a nucleotide fasta file.
There are distinct pipelines for eukaryotic and prokaryotic genomes. BUSCO decides which pipeline to use depending on the dataset selected.
Each pipeline uses a different gene predictor tool, explained below.
The predicted genes are then passed to HMMER, which scores them against the HMM profiles for the BUSCO marker genes in a given dataset.
The HMMER results are then passed through a number of filters to categorise the gene matches as Complete and Single-copy, Complete and Duplicated, Fragmented or Missing. These filters ensure that:
- no BUSCO IDs is categorised multiple times
- no gene match is assigned to multiple BUSCO IDs
- matches in which the top match is significantly better than other matches are treated as Single-copy matches
- no two gene matches overlap. If they do, the match with the lower score is discarded.
When using either Miniprot or Metaeuk, BUSCO will first examine the individual exons of a gene match to see if
each exon is matched to the HMM profile. If an exon at the flank of a gene match is not matched to the HMM profile by
HMMER, and removing this exon would resolve the overlap, BUSCO will discard the exons and update the gene coordinates
accordingly. The updated coordinates are reported in both the
full_table.tsvfile and thebusco_sequences/sequence files output. The GFF files will contain the original coordinates and also the coordinates of the exons, facilitating users to see by inspection which exons were removed.
Note: When running BUSCO using the fungi_odb10 dataset, BUSCO will recognise if the missing BUSCO marker genes match the pattern of those in parasitic-reduced genomes.
This will be reported along with a recalculated score that would be obtained by disregarding these missing genes.
BBTools
At the start of all genome mode pipelines, BUSCO will run the stats.sh script from the BBTools package to provide basic statistics on the input assembly.
The --contig_break=N option is equivalent to adding n=N to the stats.sh command (where N is an integer). This parameter is set to differentiate between contigs and scaffolds in a genome assembly.
For example, if a sequence (typically a scaffold) contains a gap of Ns that exceeds the specified contig_break value (default is 10), the sequence is interpreted as being composed of two separate contigs.
This is important to assess the quality of the genome assembly. It's possible to have a genome assembled into a few scaffolds, but with numerous gaps. Since scaffolding can sometimes introduce errors or misassemblies,
understanding the assembly's fragmentation in terms of contigs (sequences devoid of or with very short gaps) is relevant. The default value of 10 is somewhat arbitrary, but reasonable in our experience.
The resulting number of contigs/contigN50 are not used in subsequent BUSCO computations.
The --scaffold_composition option is equivalent to adding gc=scaffold_composition.txt to the stats.sh command. This writes ACGTN content per scaffold to a file.
Eukaryota
As of v5.7.0, Miniprot is the default tool for eukaryotic genome mode. Miniprot is not a gene predictor, but a gene mapper, and uses a reference protein database (provided in the BUSCO datasets) to map proteins to the genome. Miniprot is generally faster than Augustus and Metaeuk (except on fungi, in which Metaeuk is still faster), and typically yields slightly higher completion scores. However, Miniprot may underperform for highly divergent assemblies (with respect to the species in the BUSCO datasets), due to its limited sensitivity to divergent orthologs. Caution should be exercised with the reported gene predictions, as many of these may contain internal stop codons and may not be suitable for further analysis. BUSCO will report anomalies detected in the final results.
Miniprot pipeline (default for eukaryota)
The typical output of the Miniprot pipeline is as follows:
<output_folder>
├── logs
│ ├── *_err.log
│ ├── *_out.log
├── run_<dataset>
│ ├── busco_sequences
│ │ ├── fragmented_busco_sequences
│ │ ├── multi_copy_busco_sequences
│ │ └── single_copy_busco_sequences
│ ├── full_table.tsv
│ ├── hmmer_output
│ ├── miniprot_output
│ │ ├── ref.mpi
│ │ └── translated_proteins
│ ├── missing_busco_list.tsv
│ ├── short_summary.json
│ └── short_summary.txt
├── short_summary.specific.<dataset>.<output_folder>.json
└── short_summary.specific.<dataset>.<output_folder>.txt
- The
logs/folder contains a detailedbusco.logfile (with DEBUG notifications) and the capturedstderrandstdoutstreams of each third party software. - The
busco_sequences/subdirectory contains protein sequence files in FASTA format (*.faa) and GFF files (*.gff) for each BUSCO gene identified. These are subdivided into subdirectories for single-copy, multi-copy and fragmented matches. Nucleotide coding sequences are not provided in the Miniprot pipeline. This is because they can frequently contain internal stop codons and are not suitable for subsequent phylogenetic analysis. - The
full_table.tsvfile contains the complete results in a tabular format with scores and lengths of BUSCO matches, and coordinates (for genome mode) or gene/protein IDs (for transcriptome or protein modes). - The
missing_busco_list.tsvfile contains a list of missing BUSCOs. - The
short_summary.*.txtandshort_summary.*.jsonfiles contain a plain text and JSON summary of the results. - The
hmmer_outputdirectory contains tabular format HMMER output of searches with BUSCO HMMs.
Metaeuk pipeline
Metaeuk was introduced as the default gene predictor in v5.0.0, replacing Augustus. To use the Metaeuk pipeline add the --metaeuk option on the command line or set use_metaeuk=True in the config file.
Designed for eukaryotic metagenomes, Metaeuk is a fast and accurate gene predictor that uses a reference protein database (provided in the BUSCO datasets) to predict genes.
Metaeuk is faster than Augustus. It also uses less memory than Miniprot.
The Metaeuk pipeline performs an initial run and then a rerun with different parameters in an attempt to find matches for BUSCOs that were Missing or Fragmented in the first pass. The directory structure is slightly different as a result:
<output_folder>
├── logs
├── run_<dataset>
│ ├── busco_sequences
│ │ ├── fragmented_busco_sequences
│ │ ├── multi_copy_busco_sequences
│ │ └── single_copy_busco_sequences
│ ├── full_table.tsv
│ ├── hmmer_output
│ │ ├── initial_run_results
│ │ └── rerun_results
│ ├── metaeuk_output
│ │ ├── combined_nucl_seqs.fas
│ │ ├── combined_pred_proteins.fas
│ │ ├── initial_results
│ │ └── rerun_results
│ ├── missing_busco_list.tsv
│ ├── short_summary.json
│ └── short_summary.txt
├── short_summary.specific.<dataset>.<output_folder>.json
└── short_summary.specific.<dataset>.<output_folder>.txt
- For the Metaeuk pipeline, the
busco_sequences/subdirectory also contains nucleotide coding sequence files in FASTA format (*.fna).
Augustus pipeline
Augustus is a widely used gene predictor for eukaryotic genomes. It is the default gene predictor for eukaryotic genomes in BUSCO v4.0.0 and earlier.
To use the Augustus pipeline add the --augustus option on the command line or set use_augustus=True in the config file.
Augustus is a self-training gene predictor, and requires a reference file for training.
BUSCO specifies a default training set for each dataset lineage, but it is possible to specify an alternative training set using the --augustus_species option.
Augustus gene predictions are the most reliable for downstream analysis.
The Augustus pipeline includes a number of Augustus scripts. The output directory structure is built around the various steps in this procedure. BUSCO also performs a second pass to find matches for BUSCOs that were Missing or Fragmented in the first pass. The rerun parameters are obtained by training Augustus on the results found in the first pass.
<output_folder>
├── blast_db
├── logs
├── run_<dataset>
│ ├── augustus_output
│ │ ├── extracted_proteins
│ │ ├── gb
│ │ ├── gff
│ │ ├── predicted_genes_initial_run
│ │ ├── predicted_genes_rerun
│ │ ├── retraining_parameters
│ │ │ └── BUSCO_<output_folder>
│ │ └── training_set.db
│ ├── blast_output
│ │ ├── ancestral_variants_missing_and_frag_rerun
│ │ ├── coordinates.tsv
│ │ ├── coordinates_missing_and_frag_rerun.tsv
│ │ ├── sequences
│ │ ├── tblastn.tsv
│ │ └── tblastn_missing_and_frag_rerun.tsv
│ ├── busco_sequences
│ │ ├── fragmented_busco_sequences
│ │ ├── multi_copy_busco_sequences
│ │ └── single_copy_busco_sequences
│ ├── full_table.tsv
│ ├── hmmer_output
│ │ ├── initial_run_results
│ │ └── rerun_results
│ ├── missing_busco_list.tsv
│ ├── short_summary.json
│ └── short_summary.txt
├── short_summary.specific.<dataset>.<output_folder>.json
└── short_summary.specific.<dataset>.<output_folder>.txt
- The training parameters found in the
retraining_parameterssubdirectory are used in the Augustus rerun, and can also be used with Augustus for subsequent analysis if so desired. - The
blast_dbdirectory contains the BLAST database used in the Augustus pipeline. This is independent of the dataset used, so it is stored one level higher than the dataset-specific results.
Prokaryota
For prokaryotic genomes Prodigal is the default gene predictor. Prodigal predicts genes ab-initio, without any reference file. Prodigal also allows the translation table to be specified, which is useful for annotating genomes with non-standard genetic codes. BUSCO datasets for prokaryota already specify the optimal genetic code for a given clade. In cases in which multiple genetic codes are present within a single clade, BUSCO will automatically assess which translation table yields the highest coding density.
The typical output of the Prodigal pipeline is as follows:
<output_folder>
├── logs
├── prodigal_output
│ └── predicted_genes
│ └── tmp
├── run_<dataset>
│ ├── busco_sequences
│ │ ├── fragmented_busco_sequences
│ │ ├── multi_copy_busco_sequences
│ │ └── single_copy_busco_sequences
│ ├── full_table.tsv
│ ├── hmmer_output
│ ├── missing_busco_list.tsv
│ ├── short_summary.json
│ └── short_summary.txt
├── short_summary.specific.<dataset>.<output_folder>.json
└── short_summary.specific.<dataset>.<output_folder>.txt
- In cases in which Prodigal is run multiple times using different genetic codes the output of each run can be found in the
prodigal_output>predicted_genes>tmp.
Transcriptome mode
Transcriptome mode can be selected using the --mode option set to transcriptome. Depending on the dataset used, BUSCO will run either the eukaryota pipeline or the prokaryota pipeline.
Eukaryota
For eukaryotic transcriptomes BUSCO uses the Eukaryota Metaeuk pipeline, and has the same output directory structure. The input file should be a nucleotide fasta file.
Prokaryota
For prokaryotic transcriptome mode, BUSCO runs BLAST searches to find matches for the BUSCO marker genes, followed by HMMER scoring of all six frame translations.
These translations are naive, ignoring start and stop codons only in order to apply hmmsearch and do not represent proteins
The input file should be a nucleotide fasta file.
The typical output of the prokaryotic transcriptome pipeline is as follows:
<output_folder>
├── blast_db
├── logs
├── run_<dataset>
│ ├── blast_output
│ │ ├── coordinates.tsv
│ │ ├── sequences
│ │ └── tblastn.tsv
│ ├── busco_sequences
│ │ ├── fragmented_busco_sequences
│ │ ├── multi_copy_busco_sequences
│ │ └── single_copy_busco_sequences
│ ├── full_table.tsv
│ ├── hmmer_output
│ ├── missing_busco_list.tsv
│ ├── short_summary.json
│ └── short_summary.txt
├── short_summary.specific.<dataset>.<output_folder>.json
├── short_summary.specific.<dataset>.<output_folder>.txt
└── translated_proteins
Protein mode
Protein mode can be selected using the --mode option set to proteins. The input file should be a protein fasta file.
The input file is passed directly to HMMER, which scores the proteins against the HMM profiles for the BUSCO marker genes in a given dataset.
The typical output of the protein pipeline is as follows:
<output_folder>
├── logs
│ ├── busco.log
│ ├── hmmsearch_err.log
│ └── hmmsearch_out.log
├── run_<dataset>
│ ├── busco_sequences
│ │ ├── fragmented_busco_sequences
│ │ ├── multi_copy_busco_sequences
│ │ └── single_copy_busco_sequences
│ ├── full_table.tsv
│ ├── hmmer_output
│ │ ├── initial_run_results
│ │ └── rerun_results
│ ├── missing_busco_list.tsv
│ ├── short_summary.json
│ └── short_summary.txt
├── short_summary.specific.<dataset>.<output_folder>.json
└── short_summary.specific.<dataset>.<output_folder>.txt
Auto-select lineage
Automated lineage selection
Important Notice: Auto-lineage Compatibility
Some users may experience unexpected lineage assignments when running BUSCO with SEPP v4.5.6 (this may concern users who installed BUSCO v6.0.0 from Conda before Dec. 2).
This is due to a compatibility issue between the auto-lineage files and this newer version of SEPP.
The last BUSCO v6.0.0 on Conda pins SEPP to v4.5.5 by default. Make sure your installation is using SEPP 4.5.5 for auto-lineage compatibility, and reinstall from Conda if needed.
(For the Docker image, no action is required).
Since v4.0.0, it is possible to let BUSCO automatically select the dataset that is best suited to the input assembly.
This is particularly useful for metagenomic analysis, where the lineage of the input assembly may be unknown.
The Auto-Select Lineage pipeline can be invoked using the --auto-lineage option, e.g.
busco -m MODE -i INPUT -o OUTPUT --auto-lineage
This pipeline runs the respective BUSCO pipelines on the generic lineage datasets for the domains archaea, bacteria and eukaryota.
The --auto-lineage-euk and --auto-lineage-prok options run auto-placement just on eukaryota and prokaryota (archaea, bacteria) parent datasets, respectively,
and may save on computational resources if compatible with your experimental goal.
BUSCO selects the phylogenetic tree associated with the highest scoring parent dataset.
BUSCO then uses SEPP to place the input assembly on the selected tree.
The most appropriate BUSCO dataset is selected based on this placement.
BUSCO evaluations are valid when an appropriate dataset is used, i.e., the dataset belongs to the lineage of the species being analysed. Because of overlapping markers/spurious matches among domains, BUSCO matches in another domain do not necessarily mean that your genome/proteome contains sequences from this domain. However, a high BUSCO score in multiple domains might help you identify possible contaminations.
The typical output directory structure for the auto-select lineage pipeline is as follows:
<output_folder>
├── auto_lineage
│ ├── run_archaea_odb12.2
│ │ ├── busco_sequences
│ │ │ ├── fragmented_busco_sequences
│ │ │ ├── multi_copy_busco_sequences
│ │ │ └── single_copy_busco_sequences
│ │ ├── full_table.tsv
│ │ ├── hmmer_output
│ │ ├── missing_busco_list.tsv
│ │ ├── placement_files
│ │ │ ├── marker_genes.fasta
│ │ │ ├── output_alignment.fasta
│ │ │ ├── output_alignment_masked.fasta
│ │ │ ├── output_placement.json
│ │ │ └── output_rename-json.py
│ │ ├── short_summary.json
│ │ └── short_summary.txt
│ ├── run_bacteria_odb12.2
│ │ ├── busco_sequences
│ │ │ ├── fragmented_busco_sequences
│ │ │ ├── multi_copy_busco_sequences
│ │ │ └── single_copy_busco_sequences
│ │ ├── full_table.tsv
│ │ ├── hmmer_output
│ │ ├── missing_busco_list.tsv
│ │ ├── short_summary.json
│ │ └── short_summary.txt
│ └── run_eukaryota_odb12.2
│ ├── busco_sequences
│ │ ├── fragmented_busco_sequences
│ │ ├── multi_copy_busco_sequences
│ │ └── single_copy_busco_sequences
│ ├── full_table.tsv
│ ├── hmmer_output
│ ├── miniprot_output
│ │ ├── ref.mpi
│ │ └── translated_proteins
│ ├── missing_busco_list.tsv
│ ├── short_summary.json
│ └── short_summary.txt
├── logs
├── prodigal_output
│ └── predicted_genes
│ └── tmp
├── run_archaea_odb12.2 -> auto_lineage/run_archaea_odb12.2
├── run_methanococcales_odb12.2
│ ├── busco_sequences
│ │ ├── fragmented_busco_sequences
│ │ ├── multi_copy_busco_sequences
│ │ └── single_copy_busco_sequences
│ ├── full_table.tsv
│ ├── hmmer_output
│ ├── missing_busco_list.tsv
│ ├── short_summary.json
│ └── short_summary.txt
├── short_summary.generic.archaea_odb12.2.<output_folder>.json
├── short_summary.generic.archaea_odb12.2.<output_folder>.txt
├── short_summary.specific.methanococcales_odb12.2.<output_folder>.json
└── short_summary.specific.methanococcales_odb12.2.<output_folder>.txt
- The
auto-lineagefolder contains the results for the three parent datasets,archaea_odb12.2,bacteria_odb12.2, andeukaryota_odb12.2. - The
prodigal_outputdirectory is located at the top level, as these results are used for botharchaea_odb12.2andbacteria_odb12.2runs, along with any other possible subsequent prokaryotic runs. - The
run_<dataset>folders contain the results for the respective parent datasets. The final selected dataset will be at the top level of the results folder. - In the event that placement cannot be performed due to insufficient marker genes, BUSCO will report the results from the optimal parent dataset. This results folder will be soft-linked from the top level of the results folder.
- The
short_summary*files for the parent dataset ("generic") and the final selected dataset ("specific") are located at the top level of the results folder.
Batch mode
BUSCO allows you to run multiple analyses in batch mode. The input is a directory containing multiple fasta files. The output is a directory containing subdirectories named after the input files, which otherwise contain the same structure as described above.
<output_folder>
├── GCF_000091665.1_ASM9166v1_genomic.fna
│ ├── logs
│ │ ├── bbtools_err.log
│ │ ├── bbtools_out.log
│ │ ├── hmmsearch_err.log
│ │ ├── hmmsearch_out.log
│ │ ├── prodigal_err.log
│ │ └── prodigal_out.log
│ ├── prodigal_output
│ │ └── predicted_genes
│ ├── run_bacteria_odb12.2
│ │ ├── busco_sequences
│ │ ├── full_table.tsv
│ │ ├── hmmer_output
│ │ ├── missing_busco_list.tsv
│ │ ├── short_summary.json
│ │ └── short_summary.txt
│ ├── short_summary.specific.bacteria_odb12.2.GCF_000091665.1_ASM9166v1_genomic.fna.json
│ └── short_summary.specific.bacteria_odb12.2.GCF_000091665.1_ASM9166v1_genomic.fna.txt
├── GCF_000226975.2_ASM22697v3_genomic.fna
│ ├── logs
│ │ ├── bbtools_err.log
│ │ ├── bbtools_out.log
│ │ ├── hmmsearch_err.log
│ │ ├── hmmsearch_out.log
│ │ ├── prodigal_err.log
│ │ └── prodigal_out.log
│ ├── prodigal_output
│ │ └── predicted_genes
│ ├── run_bacteria_odb12.2
│ │ ├── busco_sequences
│ │ ├── full_table.tsv
│ │ ├── hmmer_output
│ │ ├── missing_busco_list.tsv
│ │ ├── short_summary.json
│ │ └── short_summary.txt
│ ├── short_summary.specific.bacteria_odb12.2.GCF_000226975.2_ASM22697v3_genomic.fna.json
│ └── short_summary.specific.bacteria_odb12.2.GCF_000226975.2_ASM22697v3_genomic.fna.txt
├── batch_summary.txt
└── logs
└── busco.log
- The primary difference is that there is a top-level
logs/directory containing thebusco.logfile, which contains the logs for all the analyses in the batch. There are separatelogs/folders for each run, containing thestderrandstdoutof each third party software for that run.
Batch mode with snakemake
An alternative batch mode script using snakemake is available here. This incorporates greater parallelization but must be run in --offline mode with the required datasets downloaded in advance.
Interpreting the results
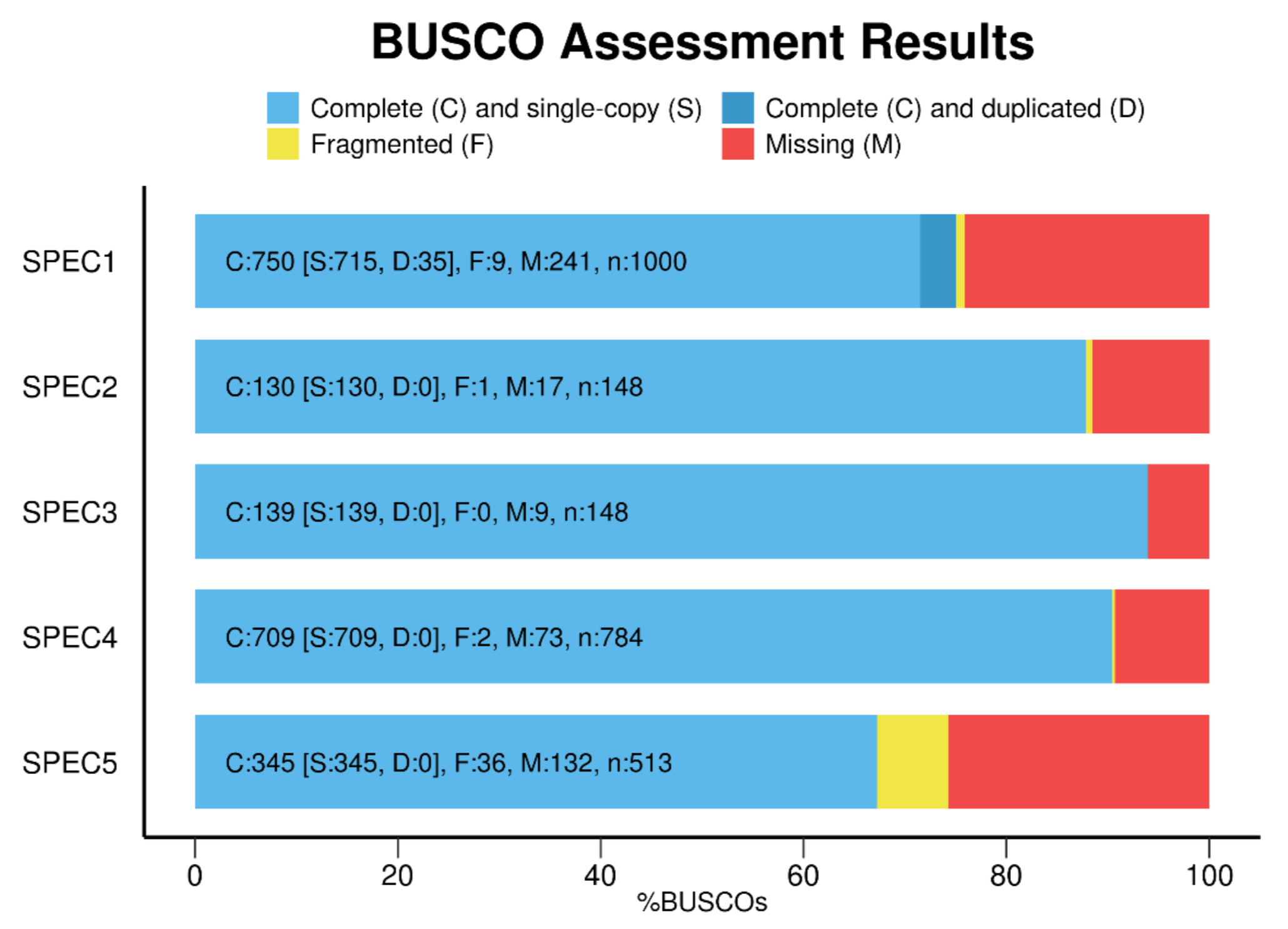
BUSCO provides a quantitative assessment of the completeness in terms of expected gene content of a genome assembly, transcriptome, or annotated gene set. The results are simplified into categories of Complete (C), Complete and single-copy (S), Complete and duplicated (D), Fragmented (F), or Missing (M) BUSCOs, where "BUSCOs" is shorthand for "BUSCO marker genes", the set of universal single-copy orthologs that are expected to be present.
Upon completion of a BUSCO run the results are displayed on screen in the following format:
---------------------------------------------------
|Results from dataset diptera_odb12.2 |
---------------------------------------------------
|C:99.1%[S:98.6%,D:0.5%],F:0.1%,M:0.8%,n:3914 |
|3878 Complete BUSCOs (C) |
|3860 Complete and single-copy BUSCOs (S) |
|18 Complete and duplicated BUSCOs (D) |
|3 Fragmented BUSCOs (F) |
|33 Missing BUSCOs (M) |
|3914 Total BUSCO groups searched |
---------------------------------------------------
When running the Miniprot pipeline an additional value E is provided, indicating the percentage of Complete matches that contain internal stop codons. This serves as a caution not to blindly use the predicted sequences for downstream analyses. Sequences predicted by Metaeuk and Augustus do not contain internal stop codons. Augustus gene predictions are recommended if you need the sequences for subsequent analysis.
-------------------------------------------------------------------------------------------
|Results from dataset eudicotyledons_odb12.2 |
-------------------------------------------------------------------------------------------
|C:97.9%[S:49.4%,D:48.5%],F:0.2%,M:1.9%,n:1990,E:8.6% |
|1948 Complete BUSCOs (C) (of which 168 contain internal stop codons) |
|983 Complete and single-copy BUSCOs (S) |
|965 Complete and duplicated BUSCOs (D) |
|4 Fragmented BUSCOs (F) |
|38 Missing BUSCOs (M) |
|1990 Total BUSCO groups searched |
-------------------------------------------------------------------------------------------
The above summary is also provided in the short summary file(s), provided in both .txt and .json formats.
These files also contain information on the dataset(s) used and the versions of all dependencies to facilitate reproducibility.
BUSCO completeness results should be interpreted in the context of the biology of the organism. Missing or duplicated genes may be of biological or technical origin. For instance, a high level of duplication may be explained by a recent whole duplication event (biological) or a chimeric assembly of haplotypes (technical). Transcriptomes and protein sets that are not filtered for isoforms will lead to a high proportion of duplicates. They should therefore be filtered before running a BUSCO analysis. Finally, focusing on specific tissues or specific life stages and conditions in a transcriptomic experiment is unlikely to produce a BUSCO-complete transcriptome. In this case, consistency across samples becomes important.
If you need help or suggestions, use the BUSCO issues board to exchange with the BUSCO development team and other users.
Complete
If found to be complete, whether single-copy or duplicated, the BUSCO matches have scored within the expected range against the BUSCO profile, and within the expected range of length alignments for the orthologous group. If in fact an ortholog is not present in the input dataset, or the ortholog is only partially present (highly fragmented), and a high-identity full-length homolog is present, it is possible that this homolog could be mistakenly identified as the complete BUSCO. The score thresholds are optimised to minimise this possibility, but it can still occur.
Duplicate matches are reported for multiple complete matches of similar quality. If a duplicate match is of significantly lower quality, for example if the score is less than 85% of the best Complete match, or if the duplicate contains internal stop codons while the other Complete matches do not, then the lower quality duplicate is discarded. This may cause the match to be reclassified as a Single-copy Complete match.
Fragmented
If found to be fragmented, the BUSCO matches have scored within the range of scores but not within the range of length alignments to the BUSCO profile. For transcriptomes or annotated gene sets this indicates incomplete transcripts or gene models. For genome assemblies this could indicate either that the gene is only partially present or that the sequence search and gene prediction steps failed to produce a full-length gene model even though the full gene could indeed be present in the assembly. When running eukaryotic datasets with the Metaeuk (default) or Augustus gene predictor, matches that produce such fragmented results are given a "second chance" with a second round of sequence searches and gene predictions with ancestral variants of the missing sequences and, in the case of Augustus, parameters trained on those BUSCOs that were found to be complete. However, this can still fail to recover the whole gene. Some fragmented BUSCOs from genome assembly assessments could therefore be complete but are just too divergent or have very complex gene structures, making them very hard to locate and predict in full.
Missing
If found to be missing, there were either no significant matches at all, or the BUSCO matches scored below the range of scores for the BUSCO profile. For transcriptomes or annotated gene sets this indicates that these orthologs are indeed missing or the transcripts or gene models are so incomplete/fragmented that they could not even meet the criteria to be considered as fragmented. For genome assemblies this could indicate either that these orthologs are indeed missing, or that the sequence search step failed to identify any significant matches, or that the gene prediction step failed to produce even a partial gene model that might have been recognised as a fragmented BUSCO match. Like for fragments, when running eukaryotic datasets with the Metaeuk (default) or Augustus pipelines, BUSCOs missing after the first round are given a "second chance" with a second round of sequence searches and gene predictions with ancestral variants of the missing sequences and, in the case of Augustus, parameters trained on those BUSCOs that were found to be complete. However, this can still fail to recover the whole gene. Some missing BUSCOs from genome assembly assessments could therefore be partially present, and even possibly (but unlikely) complete, but they are just too divergent or have very complex gene structures, making them very hard to locate and predict correctly or even partially.
Reporting BUSCO
- Report results in simple BUSCO notation:
C:89.0%[S:85.8%,D:3.2%],F:6.9%,M:4.1%,n:3023 - Use the
generate_plot.py(see below) script to produce simple graphical summaries (that are easily customisable) for your publication’s supporting online information. - Report the versions you used for all third party components. We highly recommend using the BUSCO container, whose version is sufficient to safely reproduce a run.
- Report the BUSCO set(s) you used for your assessments. Mention the creation date of the dataset, not only the name, e.g. archaea_odb12.2 (2026-05-22).
- Report the BUSCO options you used.
- Report the version(s) of the genome assembly, annotated gene set, or transcriptome that you assessed.
Plotting the results
As of v6.0.0, the plotting feature has been incorporated into the main BUSCO codebase, and the generate_plot.py script is no longer used. Plotting is done using Python and Matplotlib, replacing R and ggplot2.
To generate a plot of your BUSCO results, run
busco --plot DIRECTORY
where DIRECTORY is the directory containing your BUSCO results. You can create a separate directory for multiple results, and copy the short_summary.*.json files from each of your BUSCO runs into this directory before running the above command.
For many results, BUSCO will create plot pages to avoid an excess of results cluttering the plot.
For batch runs, pass the parent batch results directory as an argument to the --plot option.
Phylogenomics
The repository https://gitlab.com/ezlab/busco_usecases contains the script that was used to produce the phylogenomics portion of the BUSCO v3 (PMID: 29220515) paper. It has not been ported to BUSCO v4.
Troubleshooting
If you need help, please search the BUSCO issues board or open a new issue if your question has not previously been answered.
Read more >>Citation
The correct citation for version 6 of the software and the odb12.2/odb12 datasets is:
*Fredrik Tegenfeldt, Dmitry Kuznetsov, Mosè Manni, Matthew Berkeley, Evgeny M Zdobnov, Evgenia V Kriventseva, OrthoDB and BUSCO update: annotation of orthologs with wider sampling of genomes
For earlier versions of the software and datasets, please refer to the following papers:
Mosè Manni, Matthew R Berkeley, Mathieu Seppey, Felipe A Simão, Evgeny M Zdobnov, BUSCO Update: Novel and Streamlined Workflows along with Broader and Deeper Phylogenetic Coverage for Scoring of Eukaryotic, Prokaryotic, and Viral Genomes. Molecular Biology and Evolution, Volume 38, Issue 10, October 2021, Pages 4647–4654
The following protocol covers the various BUSCO running modes and workflows, BUSCO setup, guidelines to interpret the results, and additional analyses, e.g., for building phylogenomic trees and visualizing syntenies using BUSCO results:
Manni, M., Berkeley, M. R., Seppey, M., & Zdobnov, E. M. (2021). BUSCO: Assessing genomic data quality and beyond. Current Protocols, 1, e323. doi: 10.1002/cpz1.323
Citations of third-party tools
As BUSCO is reliant on a number of third-party tools, please also cite the following (depending on the pipeline used):
Miniprot
Li, H. (2023) Protein-to-genome alignment with miniprot. Bioinformatics, 39, btad014 [PMID: 36648328].
Metaeuk
Levy Karin E, Mirdita M and Soeding J. MetaEuk – sensitive, high-throughput gene discovery and annotation for large-scale eukaryotic metagenomics. Microbiome. 2020; 8:48
Augustus
Mario Stanke, Mark Diekhans, Robert Baertsch, David Haussler (2008). Using native and syntenically mapped cDNA alignments to improve de novo gene finding. Bioinformatics, 24(5), pages 637–644, doi: 10.1093/bioinformatics/btn013
Prodigal
Hyatt D, Chen GL, LoCascio PF, Land ML, Larimer FW, Hauser LJ. Prodigal: prokaryotic gene recognition and translation initiation site identification. BMC Bioinformatics. 2010 Mar 8;11:119. doi: 10.1186/1471-2105-11-119. PMID: 20211023; PMCID: PMC2848648.
HMMER
hmmer.org
Eddy SR. (2011) Accelerated Profile HMM Searches. PLoS Comput Biol 7(10): e1002195. doi: 10.1371/journal.pcbi.1002195
BLAST+
Camacho C, Coulouris G, Avagyan V, Ma N, Papadopoulos J, Bealer K, Madden TL. BLAST+: architecture and applications. BMC Bioinformatics. 2009 Dec 15;10:421. doi: 10.1186/1471-2105-10-421. PMID: 20003500; PMCID: PMC2803857.
SEPP
Mirarab S, Nguyen N, Warnow T. SEPP: SATé-enabled phylogenetic placement. Pac Symp Biocomput. 2012:247-58. PMID: 22174271; PMCID: PMC3243193.
License
The BUSCO software is licensed under the MIT License.
The BUSCO datasets are licensed under the Creative Commons Attribution-NoDerivatives 4.0 International License. To view a copy of this license, visit http://creativecommons.org/licenses/by-nd/4.0/ or send a letter to Creative Commons, PO Box 1866, Mountain View, CA 94042, USA.
Any use of these datasets for analyses in a publication or product must include the citation of the corresponding paper: https://doi.org/10.1093/molbev/msab199.